客戶文獻解讀IF=52.7:靶向SKAP2可通過調節線粒體組織和細胞骨架重塑來恢復精子活力和形態
日期:2026-01-08 16:07:00
文獻名稱:SKAP2 restores sperm motility and morphology through modulating mitochondrial organization and cytoskeletal remodeling
發表期刊:Signal Transduct Target Ther
影響因子:52.7
客戶單位:浙江大學、復旦大學
男性不育已成為全球性健康問題,其中弱畸精子癥(asthenoteratozoospermia)是常見原因之一。盡管部分病例與遺傳突變相關,但其分子機制尚不明確,且缺乏針對性治療策略。2025年12月24日,浙江大學、復旦大學研究團隊在"Signal Transduction and Targeted Therapy"期刊上發表了一篇題為" Targeting SKAP2 restores sperm motility and morphology through modulating mitochondrial organization and cytoskeletal remodeling "的研究論文。通過系統篩選與功能驗證,首次揭示了hnRNPR基因及其下游靶點SKAP2在精子形成過程中的核心調控作用,并創新性地提出基于細胞外囊泡(mEVs)的蛋白遞送策略,為男性不育的治療提供了新思路。
核心研究亮點
1. 首次揭示HNRNPR-hnRNPR-SKAP2軸在精子發生中的關鍵作用,闡明m6A依賴性剪接的新機制。
2. 突破性治療策略:利用天然細胞外囊泡遞送SKAP2蛋白,實現非侵入性修復遺傳缺陷導致的精子功能異常。
3. 臨床轉化潛力:在人類精子中驗證mEVs-SKAP2的治療效果,為弱畸形精子癥提供精準干預靶點
主要研究方法及結論
1. HNRNPR基因的突變導致弱畸精子癥及其與男性不育的關聯
研究團隊招募了572名弱畸形精子癥患者,對其中兩個家系(一個近親結婚家庭和一個非近親結婚家庭)的三名不育男性進行了全外顯子測序(WES)。結果在近親結婚家庭的兄弟二人中發現了 HNRNPR 基因的純合錯義突變(c.1540 G > A)(圖1b, c)。在非近親結婚家庭的先證者中發現了該基因的復合雜合錯義突變(c.1280 A > C, c.1369 G > A)(圖1d, e)。Sanger測序證實了這些突變在家系內與疾病表型共分離(即患者攜帶突變,父母為雜合攜帶者,健康親屬不攜帶)。突變位點的蛋白結構域定位(圖1f)鑒定出的突變均位于或鄰近hnRNPR蛋白的RGG結構域,該結構域對RNA結合功能至關重要。精子表型分析(圖1g-j)結果CASA 顯示,盡管精子濃度正常,但攜帶突變患者的精子前向運動能力和形態正常精子比例均顯著下降,符合弱畸形精子癥的臨床診斷(圖1g, h);形態學染色和TEM 揭示了精子存在廣泛的形態學異常,包括頂體脫落和頸部結構紊亂等超微結構缺陷(圖1i, j)。研究證明了 HNRNPR 基因的突變是導致弱畸形精子癥和男性不育的一個新的遺傳病因。
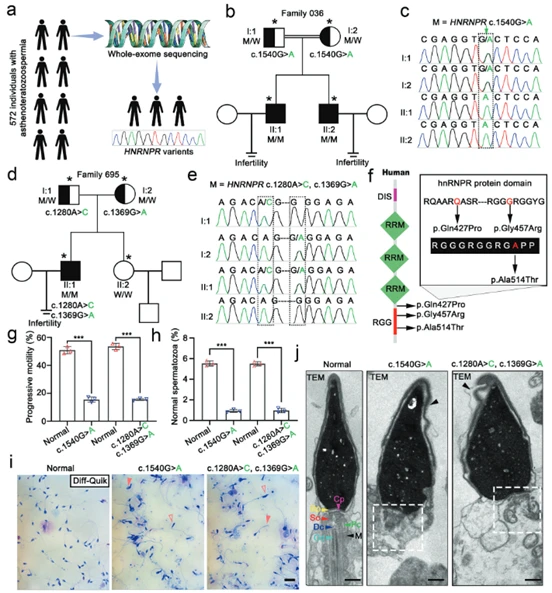
(圖1:HNRNPR基因突變導致弱畸精子癥和男性不育)
2. Hnrnpr基因突變導致小鼠弱畸精子癥和雄性不育
利用基因編輯技術,構建了攜帶Hnrnpr致病突變的基因敲入(Knock-in, KI)小鼠模型。結果發現,與野生型(WT)小鼠相比,KI小鼠的體型和睪丸與體重的比值均無顯著差異(圖2b及附圖),表明突變不影響小鼠的整體生長和睪丸大小。生育力測試顯示,Hnrnpr突變的雄性小鼠在與雌鼠合籠4個月后,完全未能產下任何后代,表現為完全不育(圖2b)。PAS染色結果顯示,KI小鼠的生精小管結構和總精子數量與WT小鼠相比沒有明顯差異(圖2c-f),這表明Hnrnpr突變不影響精子的生成數量。CASA分析結果顯示,盡管精子數量正常,但KI小鼠精子的總活力和前向運動能力均顯著低于WT小鼠(圖2g, h),復現了人類弱畸精子癥的核心特征。結果證明,Hnrnpr突變在小鼠模型中同樣導致雄性不育。這種不育并非源于精子生成障礙或數量減少,而是由于精子運動功能的嚴重缺陷,從而成功在體內再現了HNRNPR突變在人類中引起的病理表型。
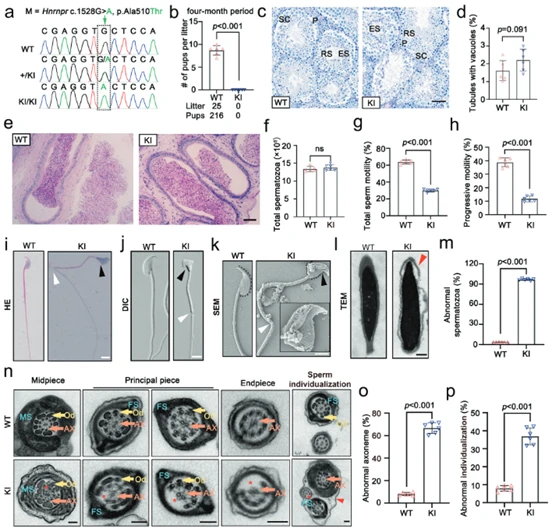
(圖2:HNRNPR基因突變導致弱畸精子癥和男性不育)
3. Hnrnpr突變導致異常精子發生和精子畸形
對野生型(WT)和突變(KI)小鼠的睪丸切片進行PAS和PNA染色,首次在精子細胞發育的第9步觀察到了明顯的頂體缺陷(圖3a),表明Hnrnpr突變在精子形成的早期階段就造成了影響。利用透射電子顯微鏡(TEM),對不同發育階段的精子細胞進行高分辨率成像,結果發現WT小鼠的頂體正常形成、遷移,細胞核正常伸長,KI小鼠表現出明顯的頂體畸形,并且發育中的頂體位置錯誤(異位),未能正確附著在細胞核表面(圖3b)。通過TEM觀察精子頭帽(一種對塑造精子頭部和鞭毛至關重要的臨時微管結構),并利用α-微管蛋白免疫染色進行定位和長度定量分析。結果發現,與WT小鼠相比,KI小鼠的精子頭帽定位異常、過度伸長且結構不對稱,其微管長度顯著長于對照組(圖3c-e)。細胞水平和超微結構水平證明,hnRNPR蛋白對于精子發生過程中的頂體正確附著、精子頭帽的正常組織以及細胞核的順利伸長至關重要。Hnrnpr突變通過破壞這些關鍵的形態建成步驟,最終導致了精子畸形。
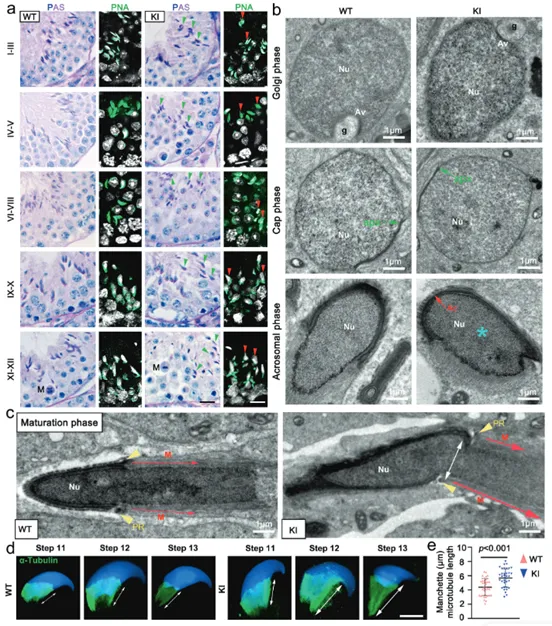
(圖3:基因敲入小鼠精子中頂體、細胞核和尾袖組裝的異常)
4. 轉錄組、蛋白質調控和RNA剪接的改變歸因于Hnrnpr突變
對野生型(WT)和突變(KI)小鼠的圓形精子細胞進行單細胞RNA測序,在KI小鼠中鑒定出498個差異表達基因(DEG),其中261個下調,237個上調(圖4a),基因本體(GO)分析顯示,這些差異基因顯著富集在與細胞骨架組織和精子運動相關的生物學通路中(圖4b),在下調基因中,Skap2基因的表達水平在KI小鼠中顯著降低(圖4c),通過RT-qPCR實驗,證實了Skap2 mRNA在突變小鼠圓形精子細胞中的表達量確實顯著下降(圖4d)。對人類和(KI/WT)小鼠的睪丸及成熟精子進行比較蛋白質組學分析,在不同物種和組織的樣本中,共鑒定到63個共同的差異表達蛋白(圖4e),這些重疊的蛋白質同樣顯著富集在精子細胞骨架組織和F-ACTIN組裝等通路中(圖4f),與轉錄組分析結果高度一致,通過整合轉錄組和蛋白質組數據,發現SKAP2是同時在RNA和蛋白質水平發生改變的關鍵分子(圖4g),在差異蛋白中,高達71.08%的蛋白與精子細胞骨架相關,其中77.15%與頂體形成和功能有關(圖4h)。熱圖進一步展示了多種細胞骨架和F-ACTIN相關蛋白的表達變化(圖4i, j)。對分離的圓形精子細胞進行蛋白質印跡分析,結果與轉錄組和蛋白質組學數據一致,SKAP2蛋白和F-ACTIN蛋白的水平在KI小鼠中均顯著降低(圖4k, l)。多組學聯合分析的結果強有力地證明了Hnrnpr突變在轉錄和翻譯兩個層面均導致SKAP2和F-ACTIN表達下調。這些分子層面的變化直接解釋了精子細胞骨架重塑缺陷,是導致精子形態異常和運動能力下降的關鍵分子機制。
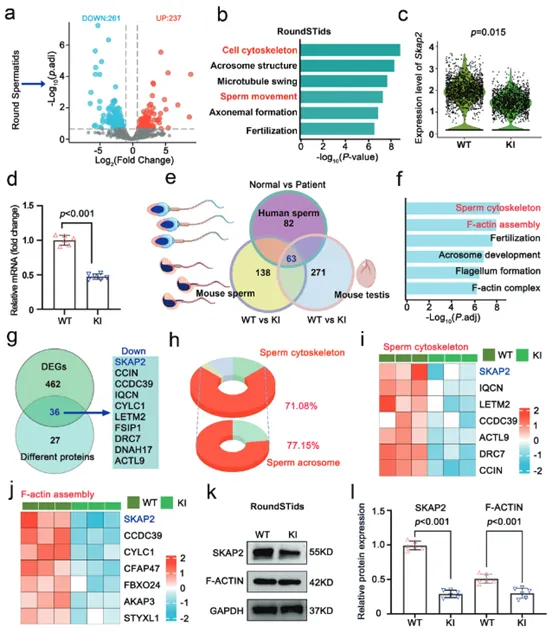
(圖4:基因敲入小鼠中異常的RNA和蛋白質特征譜)
5. hnRNPR通過m6A依賴性方式調節Skap2的可變剪接
通過RNA免疫沉淀測序(RIP-seq),鑒定hnRNPR蛋白在圓形精子細胞中直接結合的RNA靶標。發現hnRNPR蛋白結合的RNA區域,其序列特征和分布位置都與已知的m6A修飾位點高度重合(圖5a-e)。這表明hnRNPR可能作為一種m6A“閱讀器”來發揮作用。整合RIP-seq、m6A測序和差異剪接分析三組數據,發現Skap2是同時滿足“被hnRNPR結合”、“被m6A修飾”和“在突變小鼠中發生異常剪接”這三個條件的關鍵基因(圖5f)。進一步分析顯示,突變小鼠的Skap2基因發生了外顯子2的跳躍,即剪接錯誤。構建了包含Skap2基因片段的小基因報告系統,并特異性地破壞了其上的m6A修飾位點,然后在細胞中觀察剪接變化。結果發現,破壞m6A位點后,Skap2基因的外顯子2同樣發生了跳躍,成功在體外重現了突變小鼠的剪接缺陷(圖5g-k)。一系列證據鏈證明,hnRNPR通過識別Skap2 mRNA上的m6A修飾位點,來確保其被正確剪接。而Hnrnpr突變破壞了這一精確的調控過程,導致Skap2剪接異常,是其下游致病的關鍵環節。
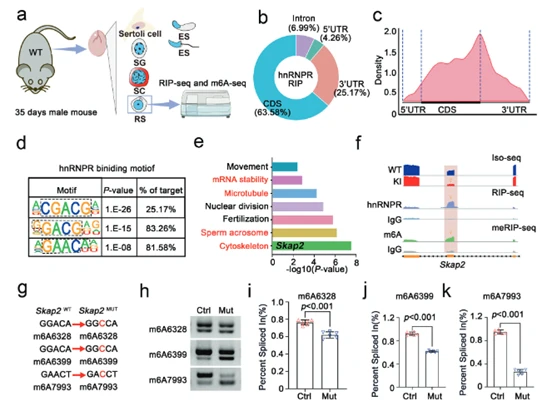
(圖5:hnRNPR蛋白通過m6A介導調控Skap2轉錄本的可變剪接)
6. SKAP2通過調節精子細胞骨架組織來維持精子發生和男性生育能力
由于全身性敲除Skap2會導致小鼠胚胎期死亡,研究人員構建了在雄性生殖細胞中特異性敲除Skap2的條件性基因敲除小鼠模型。通過RT-qPCR、蛋白質印跡和免疫熒光(IF)實驗,成功證實了SKAP2蛋白在突變小鼠的生殖細胞中被有效敲除(圖6b-e)。將基因敲除(Skap2 D-cKO)雄性小鼠與野生型雌鼠合籠,進行生育力測試,結果顯示Skap2 D-cKO雄性小鼠表現為完全不育,證實了SKAP2對雄性生育力至關重要(圖6n)。使用透射電子顯微鏡(TEM)進行精子形態學分析。觀察到大部分精子的尾部鞭毛缺乏關鍵的“9+2”軸絲微管結構,這是導致其無法正常運動的根本結構缺陷(圖6f-i)。此外,圖6l, m還顯示對精子頭部塑形至關重要的精子頭袖結構也發育異常。采用計算機輔助精子分析(CASA)技術定量檢測精子的運動參數。發現與野生型小鼠相比,Skap2 D-cKO小鼠精子的總活力和前向運動能力均顯著下降,表現出典型的弱精子癥特征(圖6j, k)。上述結果證明,在雄性生殖細胞中特異性敲除Skap2基因,足以完美復現由Hnrnpr突變導致的所有關鍵表型(包括精子尾部結構缺失、活力低下和雄性不育)。這最終確認了SKAP2是hnRNPR-SKAP2信號軸中直接導致精子異常的核心執行者。
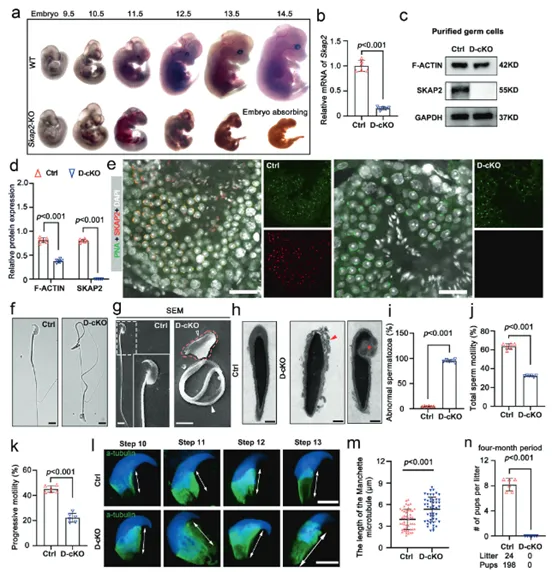
(圖6:SKAP2 缺陷干擾精子發生并導致畸形精子)
7. mEVs-SKAP2 通過改善體內的細胞骨架重塑和線粒體結構,提升精子的運動能力與形態
將裝載SKAP2蛋白的細胞外囊泡(mEVs-SKAP2),通過顯微注射方式注入Hnrnpr突變(KI)小鼠的睪丸輸出小管中,然后使用計算機輔助精子分析(CASA)評估治療效果。結果發現,與未治療組或僅注射空囊泡的對照組相比,接受mEVs-SKAP2治療的小鼠,其精子總活力和前向運動能力均顯著提升,同時形態異常的精子比例也顯著降低(圖7a-d)。利用透射電子顯微鏡(TEM)觀察治療后精子的超微結構變化,結果顯示,mEVs-SKAP2治療顯著減少了精子尾部彎曲和頂體脫落等形態缺陷,但精子尾部軸絲的“9+2”核心微管結構并未得到修復(圖7f-j),表明該療法主要改善次級結構缺陷。通過免疫熒光染色,檢測并量化精子細胞中關鍵細胞骨架蛋白F-ACTIN的組裝情況,發現mEVs-SKAP2治療能夠顯著恢復F-ACTIN的正常組裝和定位(圖7k)。這從分子層面解釋了精子形態和運動能力得到改善的原因,即SKAP2通過修復細胞骨架來發揮作用。結果證明,mEVs-SKAP2是一種有效的體內治療策略。它可以通過修復細胞骨架結構(F-ACTIN)來顯著改善精子活力和部分形態缺陷,為治療由特定基因突變引起的男性不育提供了新的潛在方案。
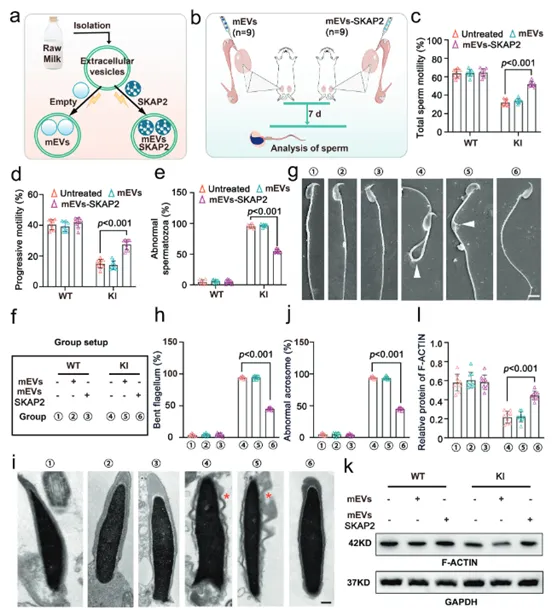
(圖7:細胞外囊泡-SKAP2恢復Hnrnp r突變小鼠的精子運動能力和形態)
采用冷凍斷裂法結合掃描電子顯微鏡技術,對小鼠精子尾部中段的線粒體進行高分辨率的超微結構觀察。在未經治療的Hnrnpr突變(KI)小鼠中,精子尾部的線粒體鞘結構紊亂且排列松散,無法形成正常的致密結構。經mEVs-SKAP2治療后,精子的線粒體結構得到了顯著改善(圖8a-d)。本研究從線粒體層面揭示了mEVs-SKAP2的治療機制。該療法能夠有效修復精子線粒體鞘的組織結構,使其排列更緊密有序。這種結構優化有助于提高ATP的生產和供應效率,從而為精子運動能力的恢復提供了堅實的能量基礎。
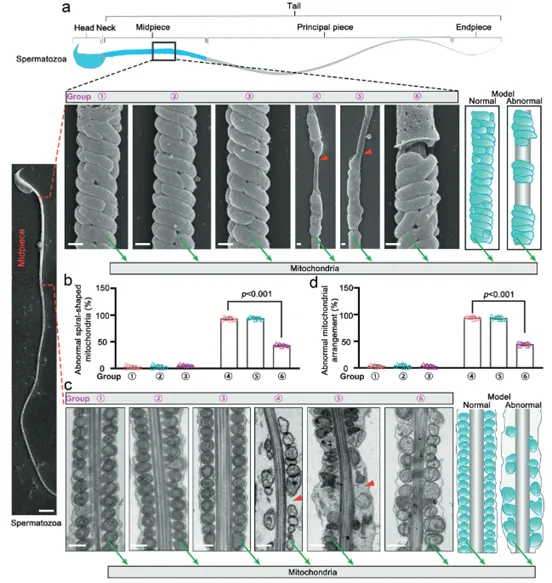
(圖8:mEVs-SKAP2恢復小鼠精子中線粒體結構)
8. mEVs-SKAP2在體外小鼠和人類模型中恢復精子活力與形態
將裝載SKAP2的細胞外囊泡(mEVs-SKAP2)與不同來源的人類精子(健康人群、HNRNPR突變患者及其他弱畸形精子癥患者)共同培養,然后使用計算機輔助精子分析(CASA)進行檢測。結果顯示,與未治療組相比,mEVs-SKAP2處理顯著增強了所有組別人類精子的總活力和前向運動能力,表明該療法在體外同樣有效(圖9b, c, e)。對上述處理后的人類精子進行形態學分析,發現mEVs-SKAP2對精子整體的形態改善效果有限,但顯著減少了精子尾部彎曲的比例(圖9d, e)。利用透射電子顯微鏡(TEM)觀察體外治療后的人類精子尾部結構,其結果與體內治療結果一致,雖然mEVs-SKAP2處理部分緩解了精子尾部彎曲,但其核心的“9+2”軸絲微管結構并未得到修復(圖9e)。同樣的結果也體現在小鼠模型的體外實驗中。通過免疫熒光染色技術,檢測體外處理后的人類和小鼠精子中關鍵細胞骨架蛋白F-ACTIN的聚合情況,發現mEVs-SKAP2處理能夠顯著促進F-ACTIN蛋白的聚合,這為體外精子運動能力的恢復提供了直接的分子機制證據(圖9f, g)。研究結果表明,mEVs-SKAP2療法在體外對人類和小鼠精子均有效。它通過重塑細胞骨架(促進F-ACTIN聚合)來顯著提升精子運動能力并改善部分形態缺陷(如尾部彎曲),但無法修復精子尾部的軸絲核心結構。這證明了該療法在臨床輔助生殖技術中具有廣闊的應用前景。
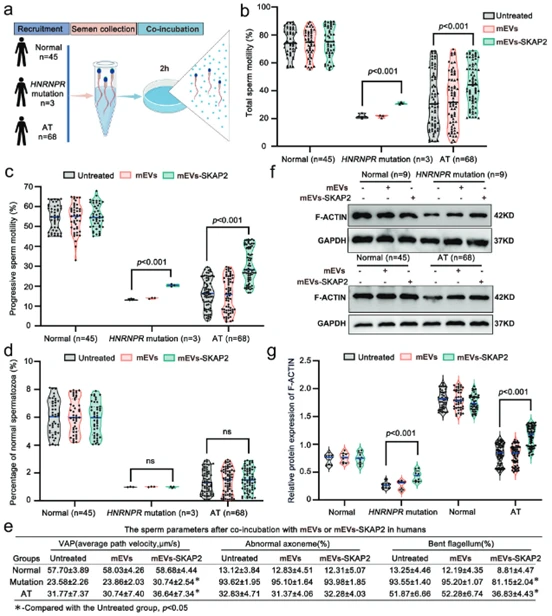
(圖9:mEVs-SKAP2緩解人類精子中的弱畸形精子癥)
研究意義
本研究揭示了由HNRNPR介導的m6A-依賴性RNA剪接調控SKAP2是導致弱畸精子癥的關鍵致病機制,并據此開發出基于細胞外囊泡的SKAP2蛋白遞送療法,為男性不育癥提供了從分子機理到靶向治療的全新策略。
CUSABIO助力產品
在本研究中,研究人員用到了CUSABIO的TUBA1A單克隆抗體(貨號:CSB-MA754656A0m),在免疫熒光實驗中檢測微管蛋白α-Tubulin的表達。

CUSABIO致力于為全球科研工作者提供讓您滿意的生物試劑,助力生命科學研究的創新與轉化。
Targeting SKAP2 restores sperm motility and morphology through modulating mitochondrial organization and cytoskeletal remodeling. Signal Transduct Target Ther, 2025 Dec 24.










